Welcome, Guest
Login/Register
How Being Poisoned Can Be Mistaken for Being Infected By ‘Germs’ and ‘Viruses’
The exposome as a missing axis of disease causation — from the fever remedy in your medicine cabinet to the outbreaks we misread as contagion
Extract:
A parent does not have to accept a single contested paradigm claim to draw a reasonable, conservative conclusion: that fever is often doing useful work, that reaching reflexively for a glutathione-depleting drug at the first sign of it is not the costless act it appears to be, and that “treat the number on the thermometer” is a medical habit, not a settled science. That is the exposome thesis made concrete, defensible, and immediately useful — and it is where this article most earns its title.
| Sayer Ji – Truth, Sovereignty, Wellness <sayerji@substack.com> Sayer Ji :May 31, 2026 : |
The Story at a Glance
The claim in one breath. Modern medicine explains illness with two stories — genes and germs.
A third, the exposome (the lifetime load of chemicals a body absorbs, including the ones we’re prescribed and injected), is real, large, and badly undercounted.
While it may not itself replace germ theory, it at least deserves to sit alongside it.
The mechanism.
Cells injured by a toxicant don’t suffer in silence — they package distress signals into tiny vesicles called exosomes and ship them through the bloodstream. Those signals can injure cells the chemical never touched. So a local poisoning can travel, and can look, to an epidemiologist, unsettlingly like contagion.
This is mainstream cell biology, shown for arsenic, cadmium, lead, air pollution, and microplastics.
The history.
Toxic outbreaks have repeatedly been misread as infections — sometimes for decades (Minamata, Itai-Itai, Toxic Oil Syndrome, the 1989 tryptophan epidemic, EVALI). The WHO and CDC now have formal protocols for telling the two apart, because the mistake is that common.
The case that matters this week. Reaching for acetaminophen at the first sign of a child’s fever rests on two assumptions — that the fever is the problem, and that the drug is free of cost. Both are shakier than they look. Fever is a coordinated defense, and the evidence that suppressing it is harmless is not established. Acetaminophen works partly by consuming glutathione, the body’s master antioxidant — exactly the reserve the immune response depends on. None of this requires any contested theory; it’s pharmacology and pediatrics. The conservative conclusion: “treat the number on the thermometer” is a habit, not settled science.
Where it gets contested.
The same lens, applied to the polio epidemic, suggests a multi-causal picture — a viral particle plus a heavy chemical load (arsenicals, DDT, medical injections) — rather than a singular pathogen. This is a serious but genuinely disputed reinterpretation, flagged as such, and nothing else in the argument depends on it.
What this is not.
This article is not focused on the claim that viruses are fake, or that all disease is toxicant-originated. It’s a claim that we built our diagnostic instruments to find pathogens, and then mistook the limits of the instrument for the limits of causation.
When we ask why people get sick, the modern answer comes in two registers: genes and germs. A disease is inherited, or it is caught. This framing organizes our research funding, our diagnostic algorithms, our public-health reflexes, and our intuitions.
It is also incomplete in a way that has become harder to defend with each passing year of toxicology research.
The missing register is the exposome —
the lifetime sum of chemical and biological exposures a body must process. The word usually conjures the environmental exposome: heavy metals, pesticides, combustion particulates, plasticizers, industrial solvents.
But it includes, just as fully, what we are given — the substances we inject, administer, and prescribe.
A compound’s having a medical purpose does not exempt it from being an exposure the body has to metabolize, sequester, or be injured by; pharmacology is a subset of toxicology, separated only by intent and dose.
Tens of thousands of synthetic chemicals are now in commerce, environmental and pharmaceutical alike; most have never been tested for their effects in combination, and many are invisible at the doses that matter.
The argument of this article is narrow and, I think, defensible: toxic exposures — encountered or introduced — can produce systemic disease that looks infectious; there is now a well-characterized molecular mechanism explaining how a localized chemical injury propagates to distant organs in a way that superficially resembles contagious spread; and our diagnostic systems were largely not built to detect any of this.
This is not a claim that opportunistic or truly pathogenic germs can’t cause disease, that viruses are purely fictional, or that infection is an illusion. It is the opposite of a grand replacement theory.
It is a claim that the exposome is a genuine, underweighted axis of causation that runs alongside the pathogen — sometimes amplifying it, sometimes mimicking it, and often hiding inside disease categories we have labeled infectious out of diagnostic habit. The most productive reframing is not “poisoned or infected.” It is the recognition that the toxic burden of the exposome shapes, amplifies, and sometimes produces from scratch the disease patterns we reflexively attribute to pathogens.
One distinction will keep this honest throughout. There is a general claim — that the exposome is a major, undercounted axis of disease causation — and there are specific claims that particular illnesses now attributed to pathogens are substantially toxic in origin.
These are not the same proposition, and they do not stand or fall together. The general claim is close to unassailable on current evidence, as you will see documented through peer-reviewed findings below. The specific reattributions vary enormously in strength:
the acetaminophen case below rests on mainstream pharmacology; the polio case is a serious but genuinely contested reinterpretation.
A reader can accept the general thesis, and the strongest specific cases, while rejecting the weaker ones — and should. This article is ordered, deliberately, from its most defensible ground to its most contested, so that nothing essential depends on the parts most easily disputed.
I. The mechanism: how a local chemical injury travels
The intuitive objection to any “toxic disease that spreads” claim is mechanical: a poison injures the cells it touches, and that should be the end of it. A chemical exposure to the liver should produce liver disease, not a whole-body syndrome, and certainly not damage in organs the chemical never reached. For most of the twentieth century, that intuition was sound enough to be load-bearing.
It is now wrong, and the thing that overturned it is one of the more important developments in cell biology of the last two decades: the discovery that cells under chemical stress actively package the molecular signatures of their injury into extracellular vesicles (EVs) — nano-scale, membrane-bound particles, the best-known subclass being exosomes— and ship them through the bloodstream to distant cells that were never exposed to the original toxicant.[^1][^2] When those vesicles are taken up by recipient cells, they can transfer the injury program itself: oxidative stress, inflammatory signaling, misfolded proteins, even the toxic metal atoms in physical cargo.
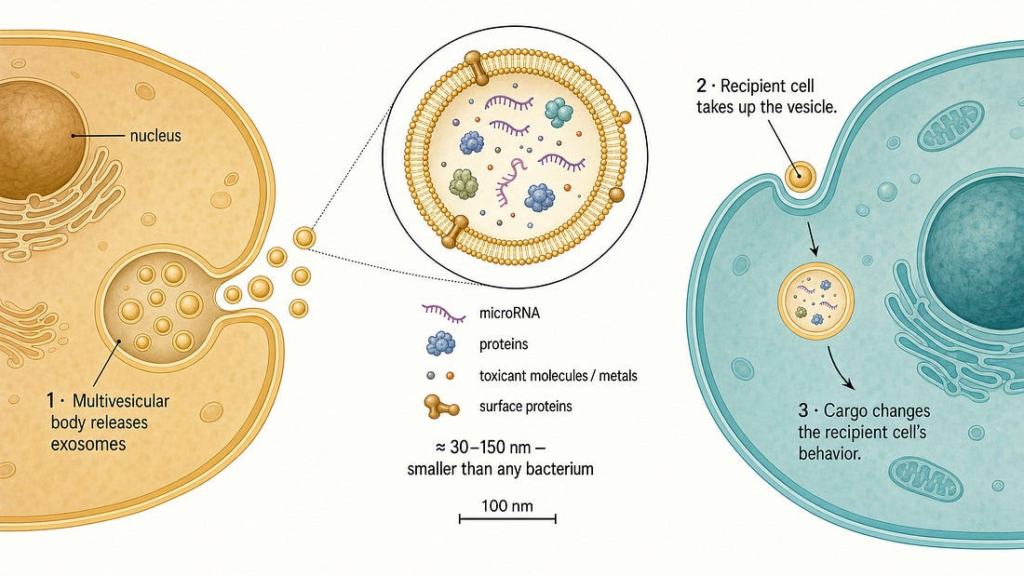
This is not a fringe mechanism. It is mainstream toxicology, and the evidence has converged from multiple independent laboratories using in-vitro models, animal studies, and human biomarker data.
The cleanest single demonstration comes from cadmium. Cadmium accumulates far more heavily in the liver than the kidney, yet it causes greater oxidative damage to the kidney — a longstanding paradox. A 2025 study resolved it: cadmium-stressed liver cells release exosomes enriched in a microRNA, miR-2137, that travels to the kidney and silences GPX4, an enzyme that suppresses ferroptotic cell death. The result is ferroptosis — a form of cell death — in an organ that was never directly exposed to the metal.[^3] Injury to a distant, unexposed organ, carried by a vesicular messenger. That is the entire thesis of this section in a single experiment.
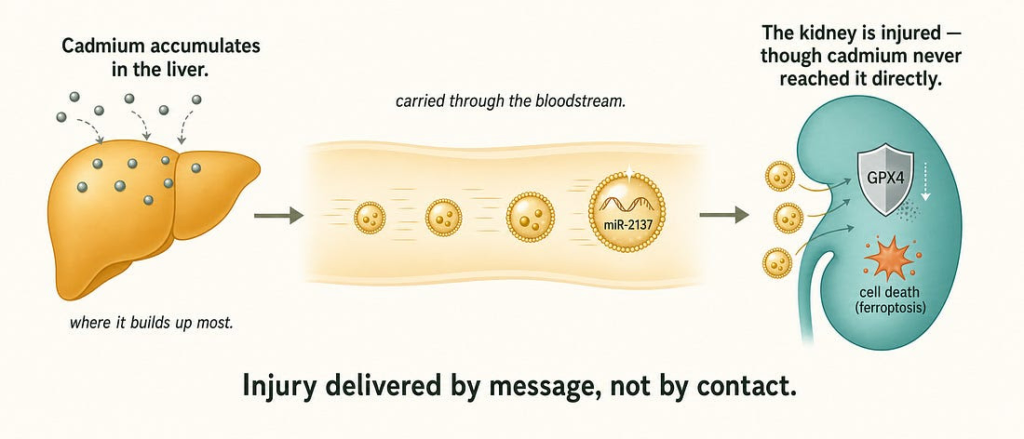
The cadmium case does not stand alone:
Lead. A 2024 study preloaded neuroblastoma cells with lead, isolated their small EVs, and applied those vesicles to lead-naïve recipient cells — producing a dose-dependent rise in intracellular lead in cells that had touched no metal, only the vesicles. The EVs were physically carrying the lead.[^4][^5]
Arsenic. A 2023 study in Toxicological Sciences supplied unusually complete proof of the principle. Bioengineered muscle exposed to a low, human-relevant dose of arsenic released EVs; when those EVs were applied to entirely arsenic-naïve muscle constructs, the naïve tissue showed markedly impaired regeneration — roughly half the recovery of contractile force — despite never contacting the metal. The result held in living mice: EVs taken from arsenic-exposed mice and injected into never-exposed mice reproduced the same impairment, with mass spectrometry identifying hundreds of differentially loaded proteins in the arsenic-EV cargo.[^6][^7] The injured tissue here is skeletal muscle rather than motor neurons, so this is proof of the mechanism — reprogrammed EVs transmitting functional injury to unexposed cells and tissue, in vivo — not a polio-specific result. But it closes the loop the principle requires.
Manganese. Manganese-exposed dopaminergic neurons release exosomes loaded with misfolded α-synuclein — the protein that aggregates in Parkinson’s disease — and those exosomes reproduce neuroinflammatory, Parkinson’s-like pathology in naïve neurons and in mice. Critically, the same exosomal markers have been detected in the blood of human welders occupationally exposed to manganese fumes, validating the animal models in living people.[^8]
Fine particulate air pollution (PM2.5). PM2.5 particles are physically carried inside macrophage-derived exosomes to unexposed recipient macrophages, where they trigger a full inflammatory cascade — NF-κB activation, TNF-α, IL-6, IL-1β. Injected into mice, the particle-loaded exosomes alone produced lung inflammation comparable to direct inhalation. The authors named the phenomenon a “secretion–particle transfer–adverse outcome chain.”[^9][^10]
Micro- and nanoplastics. The newest entry, and among the most striking. A 2024 study showed that polystyrene microplastics drive tubular kidney cells to release EVs that, applied to naïve renal fibroblasts, induced oxidative stress and fibrosis-related proteins — with the fibrotic phenotype confirmed in mice drinking water at human-relevant concentrations.[^11] A parallel finding is almost on the nose for this article’s thesis: nanoplastics internalized by brain astrocytes are exported again inside EVs, dispersing the plastic particles to neighboring cells — the vesicle acting, exactly as with lead, as a physical delivery vehicle for the toxicant itself.[^12]
If a skeptic wants the purest possible proof of concept — damage propagated to cells that received zero of the original insult — it comes from radiation biology, not chemistry. The radiation bystander effect is among the most replicated findings in the field: exosomes from irradiated cells transmit genomic instability, including DNA double-strand breaks, to cells that received no radiation, and the effect persists across more than twenty cell generations, with bystander cells in turn releasing damage-carrying vesicles to a third generation.[^13] The mechanism proposed for chemical toxicants is structurally identical to one that is already textbook for radiation.
The upshot: “spread” — injury appearing in new cells, in new organs, over time, propagating forward — is no longer a signature unique to replicating pathogens. The body’s own intercellular messaging system can carry a chemical injury in a pattern that, to an epidemiologist armed only with a case count and a clock, can look indistinguishable from contagion.
II. Toxic outbreaks misread as infection
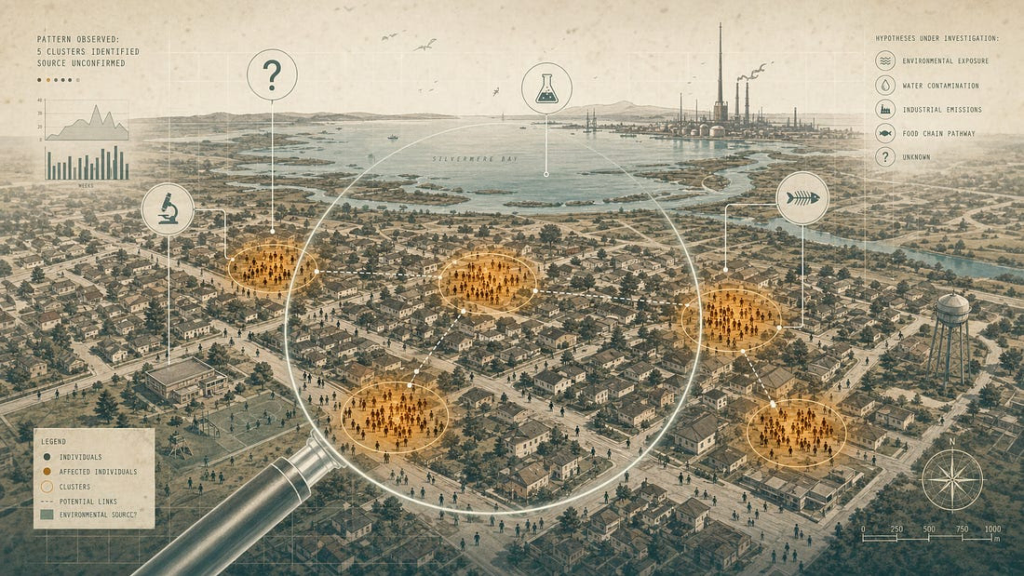
If localized chemical injury can propagate, then a shared chemical exposure across a community should be able to produce something that looks, statistically, like an infectious outbreak: clusters of cases, family aggregation, a recognizable symptom profile, a temporal wave. The historical record confirms that this happens, that it has fooled investigators for periods ranging from weeks to decades, and that the major public-health institutions formally acknowledge the problem.
The acknowledgment is explicit. In 2021 the World Health Organization published a Manual for Investigating Suspected Outbreaks of Illnesses of Possible Chemical Etiology, which states plainly that a chemical release may not be obvious and that distinguishing a non-infectious toxic cluster from an infectious outbreak requires deliberate epidemiological, toxicological, and environmental methods that standard infectious-disease investigation will not supply.[^14][^15] The CDC’s Field Epidemiology Manual similarly instructs investigators that an unusual increase in acute symptoms across a population may originate in a chemical exposure — through air, water, soil, food, or consumer products — rather than a pathogen, and provides a differential framework for telling them apart.[^16]
The documented cases span a century and four continents:
Minamata disease (Japan, 1956–1968). Methylmercury discharged into Minamata Bay produced ataxia, sensory loss, visual-field constriction, tremor, and death. The early framing was an endemic neurological disease of unknown cause, with infection among the hypotheses. It took twelve years for the government to officially attribute it to industrial mercury, by which point over two thousand were recognized as patients.[^17][^18]
Itai-itai disease (Japan). Chronic cadmium poisoning from mine tailings caused severe bone pain and fractures. The first public explanation offered, in 1955, was that the disease was caused by a microorganism. Confirmation of the cadmium cause came only after decades of contested research.[^19]
Toxic Oil Syndrome (Spain, 1981). More than twenty thousand people were affected and over three hundred died within months from rapeseed oil illegally denatured with aniline and sold as cooking oil. Because it presented as an acute lung illness and spread through communities with family clustering, it was initially investigated as a possible infectious respiratory disease.[^20]
Eosinophilia-Myalgia Syndrome (USA, 1989). Over 1,500 people developed severe myalgia, eosinophilia, and neuropathy, with 37 deaths, in a pattern of rapid geographic spread. This one is the model investigation: epidemiologists ruled out a pathogen and traced the outbreak within weeks to a contaminated batch of L-tryptophan supplement from a single manufacturer.[^21][^22]
EVALI (USA, 2019). The vaping-associated lung-injury outbreak presented as bilateral infiltrates, fever, and hypoxia — initially indistinguishable from an atypical or novel viral pneumonia. Systematic CDC investigation ruled out infection and identified vitamin E acetate, a diluent in illicit THC cartridges, as the cause.[^23]

EVALI and EMS are worth dwelling on, because they cut against any conspiratorial reading. They are cases of the system working: the infectious hypothesis was tested, found insufficient, and corrected through disciplined epidemiology. The point of this section is not that institutions are blind. It is that the toxic-versus-infectious differential is real, recurring, and difficult — difficult enough that the correct answer has sometimes taken fifty years to surface, and that an entire medical specialty, occupational and environmental medicine, exists substantially to handle it. The recurring finding from that specialty is that the most common direction of diagnostic error is toxic disease being misclassified as infectious, psychiatric, or idiopathic — not the reverse.[^24]
III. The case that matters this week: fever, acetaminophen, and the depletion of the body’s own defense

The previous sections describe a mechanism and a pattern. This one describes a decision that is being made, right now, in millions of homes — and it is the place where the exposome lens has the most immediate, actionable weight, precisely because it requires none of the contested historical claims that follow it. Everything in this section rests on mainstream pharmacology and mainstream pediatric literature. I am going to be equally careful about what it does not establish, because the stakes here are a child’s body and the wrong overcorrection is as dangerous as the status quo.
Start with the reflex.
A child spikes a fever; a parent reaches for acetaminophen (paracetamol; Tylenol). This is among the most universal acts in modern medicine — acetaminophen is the most commonly used medication in children under twelve, with one survey finding weekly use in roughly a quarter of children under two.[^25] The reflex rests on two assumptions that feel like common sense: that the fever is part of the problem, and that the drug is essentially free of cost at normal doses. Both assumptions are shakier than the reflex suggests.
What fever actually is.
Fever is not a malfunction; it is a coordinated, metabolically expensive immune response, conserved across vertebrates, that raises body temperature to impair pathogen replication and accelerate immune function. The relevant question is therefore not “how do we suppress it” but “what happens when we do.” The honest answer is that the evidence is mixed and genuinely unsettled — which is itself the point, because the reflex assumes the answer is settled in favor of suppression, and it is not. A systematic review and meta-analysis found that antipyretic treatment increased influenza mortality in animal models, with a pooled odds ratio of 1.34 — a signal worth taking seriously, though it is important to be clear that no equivalent human mortality trial has been done.[^26] A large prospective study of critically ill patients found that acetaminophen and NSAIDs were independently associated with increased mortality specifically in septic patients.[^27] Cutting the other way, a randomized controlled trial in adults with influenza found that regular paracetamol had no significant effect on viral load or clinical outcomes.[^28] The state of the evidence is not “fever suppression is proven harmful.” It is “the assumption that fever suppression is harmless is not established, and several lines of evidence suggest it may not be — while the definitive human trials have not been done.” For a near-universal intervention in children, that gap is itself the finding.
The hidden cost: glutathione. The deeper point is mechanistic, and it is not in dispute. Acetaminophen is cleared partly through a minor pathway that produces a reactive, toxic metabolite called NAPQI, which the body neutralizes by conjugating it with glutathione — the cell’s master antioxidant and a central node of its detoxification capacity.[^29] At overdose this consumes glutathione catastrophically, which is the textbook mechanism of acetaminophen liver failure. But the relevant fact for ordinary use is subtler: the depletion is not only an overdose phenomenon. Modeling and measurement indicate that chronic therapeutic dosing meaningfully lowers liver glutathione, with recovery taking days, and acetaminophen demonstrably depletes glutathione in the lung as well as the liver.[^30][^31] In other words, a drug given precisely when a child’s body is mounting an oxidatively demanding immune response also draws down the antioxidant reserve that response depends on. The two effects compound: suppress the fever, and deplete the defense, at the same moment.
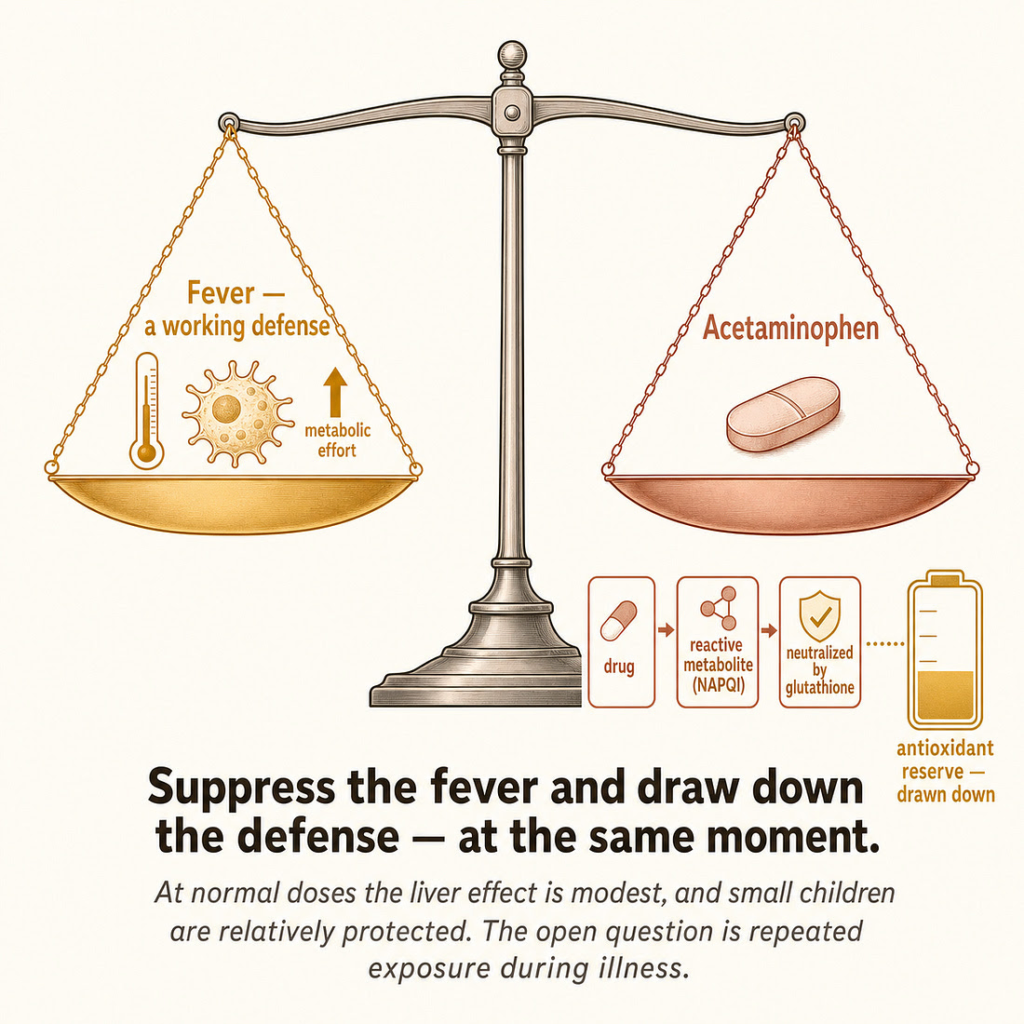
Where this stops being speculative — and where it stops, period.
his is the discipline the argument requires. The strong, defensible claims are: the glutathione-depletion mechanism is real; it operates to a degree at therapeutic and especially repeated doses; and it is plausibly more consequential in the settings where children most often receive the drug — during febrile illness, when appetite and therefore glutathione substrate are low, and in the subset of children with genetic variants (GST-null genotypes) that reduce their antioxidant capacity.[^32] The large international ISAAC study found a strong, dose-dependent association between childhood acetaminophen use and later asthma, and the proposed mechanism is exactly this glutathione/oxidative-stress pathway.[^33] That association is real and replicated.
And here is the boundary, stated plainly.
The asthma association is observational, and the standard objection is confounding — respiratory infections both prompt acetaminophen use and independently relate to asthma, so the drug could be a marker of illness rather than its cause. But confounding-adjustment is not a neutral act, and this is where the conventional reading and the toxicological one genuinely collide.
Adjusting for a true confounder removes a spurious signal; adjusting for a variable that lies on the causal pathway — a mediator — removes a real one.
The distinction is not always knowable in advance, and the choice can decide a study’s result.
The clearest live example is acetaminophen and neurodevelopment: a large 2024 Swedish cohort reported no association after extensive adjustment and sibling comparison, a result widely read as reassurance — yet critics noted the analysis recorded acetaminophen use in only ~7.5% of mothers against population exposure of 40–65%, a misclassification that biases toward the null, and that several of the adjusted covariates (inflammation, oxidative stress) are precisely the conditions a toxic mechanism would act through, making them mediators rather than confounders. An earlier study measuring acetaminophen directly in umbilical-cord blood found a dose-dependent association with later diagnoses. The point here is not to declare the question settled in either direction — it is not — but to show that “the association disappears after adjustment” can mean a confound was removed or can mean a mechanism was adjusted out of view, and that distinguishing the two is exactly the kind of analysis our pathogen-shaped, single-cause habits perform badly.
Why this is the useful core of the whole argument.
Notice what this section did not need. It did not need the claim that viruses are misidentified vesicles, or that polio was chemical, or any reinterpretation of infectious-disease history. It needed only mainstream pharmacology (NAPQI and glutathione), mainstream pediatrics (the asthma literature, the fever-outcome studies), and the willingness to notice that a reflex we treat as obviously safe has an unexamined toxic dimension — an exposome dimension — hiding in plain sight.
A parent does not have to accept a single contested paradigm claim to draw a reasonable, conservative conclusion: that fever is often doing useful work, that reaching reflexively for a glutathione-depleting drug at the first sign of it is not the costless act it appears to be, and that “treat the number on the thermometer” is a medical habit, not a settled science. That is the exposome thesis made concrete, defensible, and immediately useful — and it is where this article most earns its title.
IV. A contested case: polio as a multi-causal phenomenon
The acetaminophen section stands on mainstream ground. This one does not, and I want to mark the change in footing honestly, because it is exactly the kind of claim that — if it fails — a reader will use to discard everything before it. So treat what follows as a harder, more contested application of the same lens, offered not as proof but as the most serious version of an argument that has a long heterodox history and a real evidentiary core. If you find it unpersuasive, the preceding sections stand entirely without it.
The toxic-cofactor reading of polio is not new, and intellectual honesty requires saying so rather than presenting it as fresh discovery. It has a heterodox lineage running from the early 1950s to the present, and that lineage is part of why the thesis is treated as fringe — some versions of it overreach badly, into outright denial that poliovirus causes disease.[^35] The version defended here is deliberately narrower than its predecessors: it keeps the virus as a genuine and necessary cause, and is built from the primary clinical and toxicological record rather than from the incidence-correlation graphs that the older literature leaned on. Where that literature says “pesticides, not virus,” this section says “virus and exposome, disaggregated” — a materially different and more defensible claim.
No disease illustrates the cost of single-pathogen thinking more sharply than paralytic poliomyelitis. The claim is narrower and fully sourced to mainstream literature — that “polio,” as historically counted, was a composite of causes, of which the virus while considered necessary from the mainstream perspective is insufficient alone according to our thesis, and that a heavy concurrent exposome load shaped who among the infected was actually paralyzed. That load was both environmental and introduced: the arsenicals physicians prescribed, the DDT sprayed on children, the injections and surgeries administered during viral circulation, and — later — the live vaccine itself are all exposures the body had to process, and all belong in the causal account.
The category was never one pathogen.
Before 1958, “polio” was a clinical diagnosis — flaccid paralysis, no virological confirmation required. When 1,060 suspected cases in a 1958 Michigan epidemic were finally subjected to virological testing, more than half yielded no poliovirus at all; Coxsackie and ECHO viruses accounted for more of the “non-paralytic poliomyelitis” and “aseptic meningitis” cases than poliovirus did, and many paralytic patients had no identifiable viral cause.[^34] Whatever was being counted as polio, it was not a clean single-pathogen tally.
The 1955 reclassification.
In 1955 — the year the Salk vaccine launched — the diagnostic threshold tightened: paralysis now had to persist and be confirmed at 60 days, and other enteroviruses had to be laboratory-excluded. Dr. Bernard Greenberg, head of biostatistics at the UNC School of Public Health and chair of the American Public Health Association’s Committee on Evaluation and Standards, testified to Congress in 1962 that the change in diagnostic criteria alone predetermined a decline in counted paralytic cases in 1955–1957, vaccine or no vaccine.[^36] This does not erase a real vaccine effect; it means the post-1955 statistical drop conflates a genuine intervention with a definitional artifact, and the two have rarely been disaggregated in the popular narrative.
Chemicals produce paralysis identical to polio.
This is the part most likely to surprise, and it is not a fringe claim. A 1984 review in Reviews of Infectious Diseases — published under the Infectious Diseases Society of America — states that chemical poisons including arsenic, triorthocresyl phosphate, and organophosphorus insecticides were responsible for paralysis affecting groups of people.[^37]
The mechanism is selective destruction of anterior horn motor neurons, the same cell population poliovirus targets, producing a clinically and pathologically overlapping flaccid paralysis. Arsenic neuropathy mimicking Guillain-Barré has been documented from a 1987 case series through 2025.[^38][^39] As early as 1908, a Massachusetts State Board of Health report concluded that metallic poisons including arsenic produced spinal-cord lesions that did not differ from those of anterior poliomyelitis.[^40] Arsenic exposure was not rare in the epidemic era: arsenical pharmaceuticals were AMA-endorsed, lead-arsenate was sprayed on produce, and arsenic pigments were in wide use.
DDT: a real correlation that must be stated with its caution.
DDT was sprayed on children and at public venues as a supposed anti-polio measure in the 1940s and early 1950s, despite a lack of evidence it helped — a history documented in a 2017 peer-reviewed paper in Environmental History.[^41] That DDT is a serious neurotoxin was not a fringe view at the time: a 1955 JAMA editorial classified it as acting primarily on the central nervous system, producing — in its own list — hyperexcitation, tremors, “spastic or flaccid paralysis,” and convulsions.[^42] And DDT plugs into the vesicular mechanism of Section I: a 2017 paper links DDT to extracellular-vesicle biogenesis directly, proposing that the organochlorine raises neurodegeneration risk by inducing the vesicular budding that propagates misfolded proteins between neurons.[^43] That paper is a mechanistic hypothesis built on the authors’ bud-formation work, not proof of a DDT-to-paralysis exosomal chain — but it removes any claim that DDT sits outside the EV story; it does not.
Two cautions still belong in the same breath as the JAMA quote, and dropping either is how the claim tips into overreach. First, scope: DDT’s characteristic toxicity is hyperexcitatory — tremor and convulsion-dominant — and the editorial names the central nervous system broadly, not the anterior horn specifically; the clean, selective anterior-horn destruction that pathologically mimics polio belongs to arsenic and the organophosphates discussed above, not to DDT. Second, causation: the overlap between peak DDT use and peak polio incidence (and between its phase-out and the decline) is a real correlation, but the Salk vaccine was deployed in the same window, and timeline alone cannot separate them. The most defensible statement by mainstreame standards is that DDT plausibly amplified neurological vulnerability during the epidemic era — not that it produced polio-like flaccid paralysis by the poliovirus mechanism, and not that it was the primary driver of the epidemics.
That said, Dissolving Illusions: Disease, Vaccines, and The Forgotten History by Suzanne Humphries MD (, Roman Bystrianyk should be read to deepen one’s understanding of the possibility that the exposome and the live polio vaccines themselves were and still are the primary driver of today’s polio outbreaks.
Provocation poliomyelitis: the injection as an exposome event.
Independent of any chemical claim, it is established clinical history that routine medical interventions raised paralysis risk — the clearest case of an introduced exposure shaping a disease blamed wholly on a pathogen. Tonsillectomy during poliovirus circulation was linked to bulbar polio beginning in 1910 and confirmed repeatedly since.[^44][^45] The mechanism for injection-provoked paralysis was worked out in a 1998 Journal of Virology paper: skeletal-muscle injury opens a route for retrograde axonal transport of poliovirus directly into the spinal cord, facilitating central-nervous-system invasion — and the authors noted the effect still causes childhood paralysis where unnecessary injections are given during poliovirus circulation.[^46] The epidemiology matches the mechanism: a 1995 New England Journal of Medicine study of vaccine-associated polio in Romania found that children given intramuscular injections within 30 days before onset had an odds ratio of 31.2 for paralytic disease — a more than thirty-fold increase.[^47] An injection is not inert. During viral circulation it was, for some children, the precipitating exposure.

The virus is mostly commensal.
The WHO states roughly 1 in 200 poliovirus infections leads to any paralysis; the ECDC notes about 70% are entirely asymptomatic.[^48][^49]
The decisive variable is therefore the host and its conditions, not mere exposure.
The starkest illustration is a natural experiment: the remote Brazilian Xavante had antibodies to all three poliovirus serotypes in nearly every tested individual — and zero cases of paralytic polio.[^41] Universal infection, no paralysis. What determines paralysis among the infected, per peer-reviewed work, includes innate-immune genetic variants, nutritional status, prior injection or tonsillectomy, age at exposure, and concurrent toxic load — precisely an exposome story.[^50][^37]
The vaccine as an introduced exposure.
A live vaccine is, by definition, a biological agent introduced into the body — and the polio vaccines carried documented exposure risks of their own, quite apart from their benefit. The first is the vaccine virus itself: the live oral vaccine (OPV) is genetically unstable and can revert to neurovirulence with as few as two nucleotide mutations, which is why the United States discontinued OPV in 2000.[^51] By 2022, more global paralytic cases were caused by vaccine-derived strains than by wild virus — a fact the WHO openly builds its “OPV endgame” strategy around.[^52][^48] In India, peer-reviewed surveillance by the Puliyel group found non-polio acute flaccid paralysis running far above the expected background rate and correlating with the number of OPV doses administered per region, with rates declining when dosing was reduced after 2012.[^53][^54]
The caution belongs in the text:
India’s unusually aggressive surveillance inflates raw rates relative to other countries — a legitimate objection — but it does not by itself explain the within-country, dose-response gradient or the temporal decline when dosing dropped, and the finding has not been formally refuted in the literature.
The second risk was a contaminant.
Between 1955 and 1963, a substantial fraction of polio vaccine was contaminated with Simian Virus 40 (SV40), a monkey virus carried over from the cell cultures used in manufacture, and tens of millions of people were exposed before the contamination was identified and cleared.[^55] SV40 is oncogenic in animal models, and its DNA has since been reported in several human tumor types — but the epidemiology of human cancer risk has not borne out a clear population-level signal. The National Academies, reviewing the question in 2002, concluded that the evidence was inadequate to either accept or reject a causal link, while acknowledging a strong biological basis for concern.[^56] This is the honest status: an introduced biological contaminant, real and documented, whose downstream human consequence remains an open question rather than a demonstrated harm. It belongs in an exposome account precisely as that — an exposure, logged and unresolved, not a verdict.
The adjuvant as an introduced exposure.
The principle generalizes beyond polio to the broader vaccine pharmacopeia, and the clearest worked example is the aluminum adjuvant used in many non-live vaccines — a case where the mechanism is well characterized even where downstream clinical consequences remain unsettled.
Aluminum hydroxide is a deliberate immune irritant, and the molecular path by which it acts is now mapped. It activates the NLRP3 inflammasome — the same sterile-inflammation sensor triggered by the body’s most potent danger signals — driving release of IL-1β and IL-18 in an NLRP3- and ASC-dependent manner.[^57] NLRP3 activation in turn drives the secretion of ASC-laden extracellular vesicles, and those vesicles propagate the inflammasome-activation state to naïve recipient cells: EVs carrying inflammasome cargo activate NF-κB signaling in cells that were never exposed to the original trigger, whereas vesicles from unactivated cells do not.[^58] This is the same EV-mediated signal propagation documented for the chemical toxicants of Section I, arising here from an injected compound. And aluminum does not simply clear: adjuvant particles are phagocytosed by macrophages that biopersist and can traffic the metal through lymphatics toward the brain along a monocyte-chemoattractant pathway, with injection-site aluminum deposits documented years after administration.[^59] None of this, on its own, establishes a particular disease — the mechanism is what is established: an introduced adjuvant activating an inflammasome, propagating that activation cell-to-cell via vesicles, and distributing through the body in cells rather than being promptly excreted. Stated at that level, it is simply another, well-characterized instance of the exposome’s central mechanic — a local exposure broadcasting its signal beyond the cells it first touched.
Assembled, these are not directly a refutation of polio virology. They are a fuller causal picture: a largely commensal virus, necessary but not sufficient for paralysis in the overwhelming majority of cases; a disease burden amplified by a concurrent exposome — environmental and introduced alike — of arsenicals, DDT, nutritional depletion, medical provocation, and eventually the vaccine’s own viral risks. Polio was analyzed for decades through a single-pathogen lens. Standard environmental epidemiology, applied in retrospect, reveals a multi-causal phenomenon.
The pattern is not unique to polio.
The most general version of this point is visible in mortality data that long predate any vaccine.
Deaths from measles, whooping cough, scarlet fever, and diphtheria fell by 90 percent or more in the United States and United Kingdom before the respective vaccines were introduced — a decline that demographers attribute to improved nutrition, sanitation, housing, and supportive care rather than to immunization.[^60]
The real mortality collapse that anchors so much of the popular narrative was largely accomplished by changing the conditions in which people lived — which is to say, by changing the exposome in its broadest sense, the total environmental load a body must withstand. The single-pathogen lens credits the pathogen’s removal; the fuller picture credits the terrain. Both matter. Only one has been counted by the medical establishment.
V. Why the blind spot persists

The exposome’s invisibility in our causal accounting is not the product of a conspiracy. It is the product of structure, and the structure is worth naming because it explains why correction is slow.
Our diagnostic tools are pathogen-shaped. A PCR assay, a culture, a serology panel — these instruments are built to find an organism, and an instrument built to find an organism will tend to return an organism or a blank, not a chemical. When the blank comes back, “idiopathic” is filed more readily than “we tested for the wrong category of cause.”
Chemicals are also genuinely harder to see. A pathogen is a discrete agent that can be isolated and named. The exposome is a diffuse, low-dose, lifelong mixture, and its most consequential effects may be synergistic — the combined action of compounds that are individually below their no-effect thresholds. These mixture effects, sometimes called the problem of “ghost molecules,” are almost never tested, because the regulatory and toxicological apparatus evaluates chemicals one at a time, each assigned its own threshold as though it acted alone. A body, by contrast, experiences them all at once, and the interactions can be more than additive: a compound that depletes glutathione lowers the threshold at which an unrelated oxidative toxicant causes harm, so two exposures each individually “safe” combine into one that is not. The regulatory regime has almost no machinery for detecting this, because testing every pairwise and higher-order combination of tens of thousands of chemicals is combinatorially impossible — which means the interactions are not so much judged safe as never examined at all.
Funding compounds the bias. Large trials are financed by sponsors who can own the result. A pathogen invites a patentable countermeasure; a diffuse environmental exposure invites expensive regulation and uncompensated remediation. The economic gradient runs steeply toward the infectious framing and away from the toxic one.
And underneath all of it sits an epistemic habit inherited from microbiology: one cause, one disease. It was a spectacularly misleading simplification. It is also the reason a multi-causal, exposure-weighted picture of illness reads as heterodox even when every individual claim in it is mainstream.
Conclusion

The phrase I have used in earlier pieces — “poisoned or infected?” — is the perhaps the wrong question, because it preserves the very either/or that has kept the exposome out of our causal accounting. The body does not experience pathogens and chemicals in separate compartments.
They act together — a virus or exosome made more dangerous by an arsenical (or intentionally weaponized via gain-of-function research), a lung primed for pneumonia by particulate load, a paralysis that required both a commensal enterovirus and a toxic threshold to cross.
What the evidence supports is that the exposome is a co-author of the disease patterns we have spent a century attributing to pathogens alone. Localized chemical injury can travel through the body’s own vesicular messaging and mimic spread; shared exposures can mimic outbreaks well enough to fool investigators for decades. And the lens is not only historical — it reaches into the medicine cabinet, where a reflexive remedy for childhood fever turns out to suppress a functional defense and draw down the body’s antioxidant reserve at the same moment, with population-scale consequences we have barely begun to study. That last case asks nothing of the reader but mainstream pharmacology and a willingness to question a habit.
The practical implication is not alarm. It is instrumentation — and, in the meantime, humility about reflexes we have mistaken for settled science.
We built a diagnostic and epidemiological apparatus exquisitely tuned to find singular pathogen (even in the absence of the fulfilment of the Koch postulates), and then mistook the limits of the instrument for the limits of causation. A medicine that could actually see the exposome — that tested chemicals in the mixtures bodies meet them in, that ran the toxic differential as reflexively as the infectious one, that paused before suppressing a fever it had never proven was harmless — would not be a medicine that had abandoned germ theory*. It would be a medicine that had finally stopped looking for its keys only under the streetlight.
*Germ theory would preserve a modicum of it credibility if it acknowledged the microbiome and the tendency towards the overgrowth of normally neutral or beneficial microorganisms or biological microvessicles in dysbiosis — often driven by the exposome or conventional interventions themselves — leading to “pathogenic” expression, instead of demonizing aspects of our micro- and macro-ecological/holobiont constitution itself.
Sayer Ji’s Substack is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.